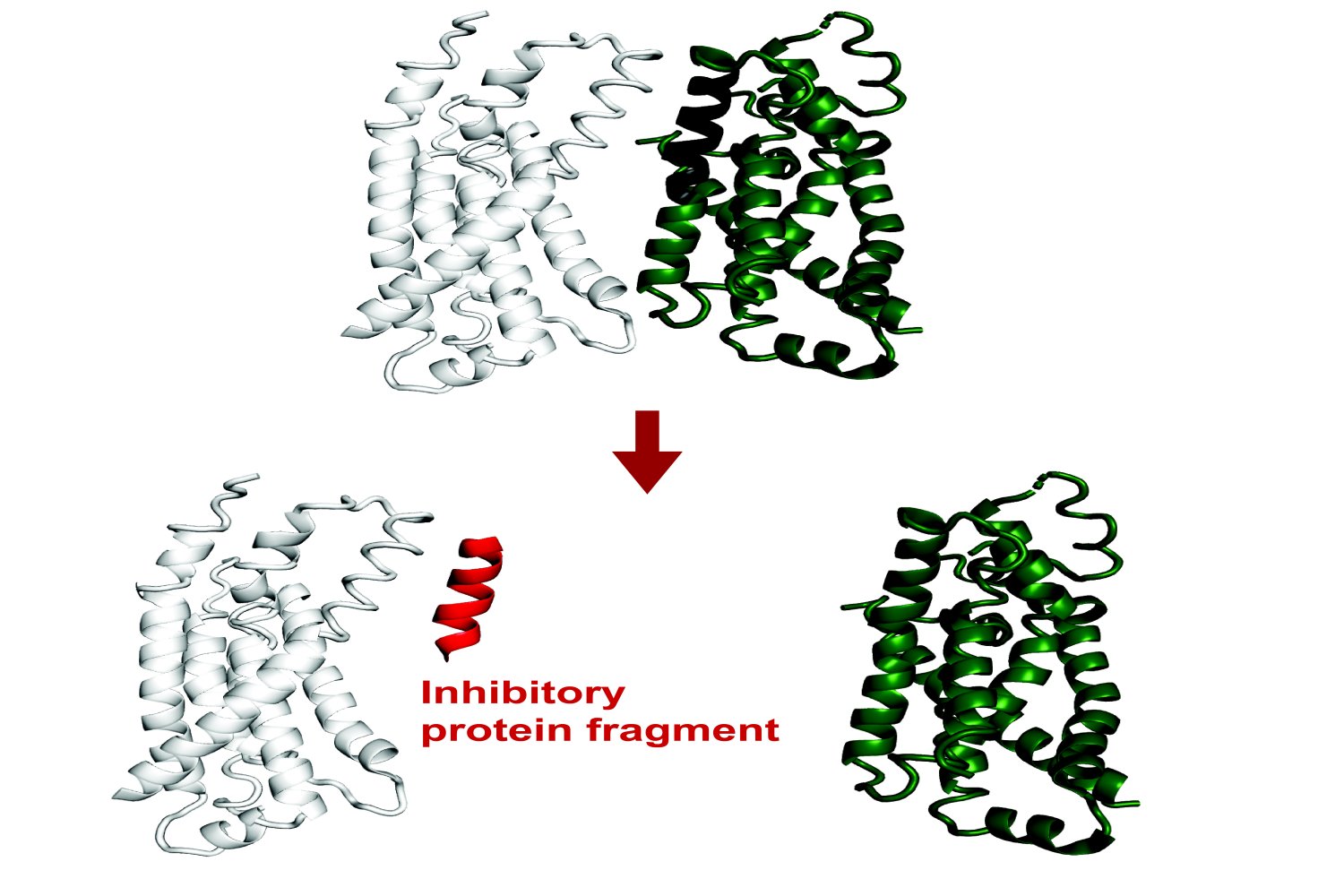
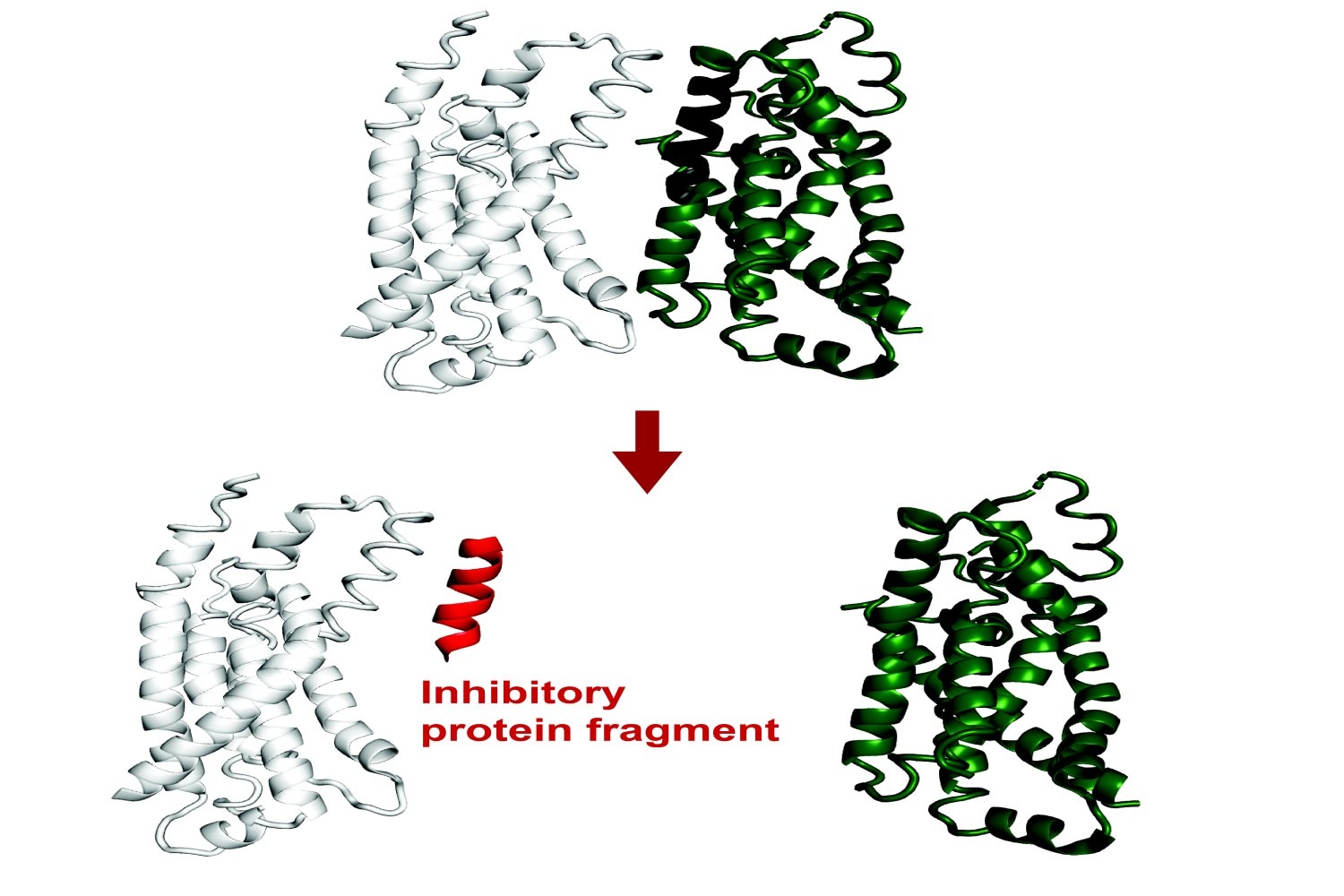
The whole biological function depends on how different proteins interact with each other. Protein-Protein interactions facilitate everything, from the transcription of DNA and the control of cell division to higher level functions in complex organisms.
However, it is not clear, however, how these functions are orchestrated at the molecular level and how proteins interact with each other – either with other proteins or with copies of themselves.
Recent results have revealed that small protein fragments have a lot of functional potential. Even if they are incomplete documents, short stretching of amino acids can always be binded at the interfaces of a target protein, summarizing native interactions. Thanks to this process, they can change the function of this protein or disturb its interactions with other proteins.
Protein fragments could therefore empower both basic research on protein interactions and cellular processes, and could potentially have therapeutic applications.
Recently published in Proceedings of the National Academy of SciencesA new method developed in the Biology Department is based on existing artificial intelligence models to predict in calculation of protein fragments which can be binded and inhibit full length proteins in E. coli. Theoretically, this tool could lead to genetically codable inhibitors against any protein.
The work was done in the laboratory of the associate professor of biology and the investigator of Howard Hughes Medical Institute Gene-wei li In collaboration with the laboratory of Jay A. Stein (1968) Professor of biology, professor of biological engineering and head of the department Amy Keating.
Take advantage of automatic learning
The program, called Fragfold, exploits Alphafold, a model of AI which has led to phenomenal progress in biology in recent years because of its ability to predict protein and protein interactions.
The goal of the project was to predict the inhibitors of the fragments, which is a new application of Alphafold. The researchers in this project have experienced experimentally that more than half of Fragfold's predictions for the connection or inhibition were precise, even when the researchers did not have previous structural data on the mechanisms of these interactions.
“Our results suggest that this is a generalizable approach to find liaison methods that are likely to inhibit the protein function, including for new protein targets, and you can use these predictions as a starting point for other experiences”, explains Co-STST and the corresponding author Andrew Savinov, a post-doc in the LI laboratory. “We can really apply this to proteins without known, without known interactions, without known interactions, Without even known structures, and we can put a little credibility in these models that we develop. ”
An example is FTSZ, a protein that is the key to cell division. It is well studied but contains an intrinsically disorderly region and, therefore, particularly difficult to study. Disordered proteins are dynamic and their functional interactions are most likely ephemeral – occurring so briefly that current structural biology tools cannot enter a single structure or interaction.
The researchers exploited Fragfold to explore the activity of FTSZ fragments, including the fragments of the intrinsically disorderly region, to identify several new connection interactions with various proteins. This jump in understanding confirms and widens the previous experiences measuring the biological activity of FTSZ.
This progression is partly important because it was made without resolving the structure of the disorderly region and because it presents the potential power of Fragfold.
“This is an example of how Alphafold fundamentally changes how we can study molecular and cellular biology,” explains Keating. “The creative applications of AI methods, such as our work on Fragfold, open up unexpected capacities and new research orientations.”
Inhibition, and beyond
The researchers accomplished these predictions by fragmenting by calculation each protein, then by modeling how these fragments would bind to the interaction partners which they thought were relevant.
They compared the cards of the predicted bond through the entire sequence to the effects of these same fragments in living cells, determined using high speed experimental measures in which millions of cells each produce a type of protein fragment.
Alphafold uses co-evolutionary information to predict the folding and generally assess the evolutionary history of proteins using something called multiple sequence alignments for each prediction execution. MSAs are essential, but are a bottleneck for large -scale predictions – they can take prohibitive time and calculation power.
For Fragfold, the researchers rather pre-populated the MSA for a full length protein once and used this result to guide the predictions for each fragment of this full length protein.
Savinov, with the former Keating Lab, Sebastian Swanson Phd '23, predicted inhibitory fragments of a diversified protein set in addition to FTSZ. Among the interactions they explored, there was a complex between the transport proteins of lipopolysaccharides LPTF and LPTG. A fragment of LPTG proteins inhibited this interaction, probably disturbing the delivery of lipopolysaccharide, which is a crucial component of the E. coli External cell membrane essential for cell shape.
“The great surprise was that we can predict the connection with such a high precision and, in fact, often predict the connection which corresponds to inhibition,” explains Savinov. “For each protein we have examined, we were able to find inhibitors.”
The researchers initially focused on protein fragments as an inhibitors because if a fragment could block an essential function in cells is a relatively simple result to measure systematically. In the meantime, Savinov is also interested in exploring the function of fragments outside of inhibition, such as fragments which can stabilize the protein to which they bind, improve or modify its function or trigger protein degradation.
Design, in principle
This research is a starting point for developing a systemic understanding of cellular design principles and what elements of deep learning models can rely to make specific predictions.
“There is a wider and more in -depth objective than we build,” explains Savinov. “Now that we can predict them, can we use the data that we have of predictions and experiences to withdraw the salient characteristics to determine what Alphafold really learned about what makes a good inhibitor?”
Savinov and collaborators also deepened how protein fragments bind, exploring other protein interactions and mutating specific residues to see how these interactions change how the fragment interacts with its target.
Examining experimentally the behavior of thousands of fragments transferred in cells, an approach known as deep mutational scanning, revealed key amino acids that are responsible for inhibition. In some cases, mutated fragments were even more powerful inhibitors than their natural and full length sequences.
“Unlike previous methods, we are not limited to identifying fragments in experimental structural data,” says Swanson. “The central force of this work is the interaction between high -speed experimental inhibition data and the planned structural models: experimental data guide us towards particularly interesting fragments, while the predicted structural models by Fragfold provide a specific and testable hypothesis on the functioning of fragments at a molecular level.”
Savinov is enthusiastic about the future of this approach and his myriad of requests.
“By creating compact and genetically codable binders, Fragfold opens a wide range of possibilities to manipulate the function of proteins”, accepts Li. “We can imagine providing functionalized fragments that can modify native proteins, modify their subcellular location and even reprogram them to create new tools to study cellular biology and treat disease.”