The process of discovering molecules that have the properties necessary to create new drugs and new materials are heavy and expensive, consuming large calculation resources and months of human work to reduce the enormous space of potential candidates.
The models of large languages (LLM) as Chatgpt could rationalize this process, but allowing an LLM to understand and reason on the atoms and the bonds that form a molecule, in the same way as the words that form sentences, have presented a scientific stumbling block.
Researchers from MIT and Mit-ibm Watson AI Lab have created a promising approach that increases an LLM with other machine learning models called models based on graphics, which are specially designed to generate and predict molecular structures.
Their method uses a basic LLM to interpret requests in natural language specifying the desired molecular properties. It automatically rocks between the LLM base and the AI modules based on graphics to design the molecule, explain the justification and generate a step -by -step plan to synthesize it. It intersects from the text, the graphic and the generation of a summary stage, combining words, graphics and reactions in a common vocabulary so that the LLM consumes.
Compared to existing LLM -based approaches, this multimodal technique has generated molecules that better corresponded to user specifications and were more likely to have a valid synthesis plan, improving the success ratio from 5% to 35%.
It has also outperformed the LLM which are more than 10 times its size and that the design molecules and the synthetic pathways only with representations based on the text, suggesting that multimodality is the key to the success of the new system.
“We hope to be an end -to -end solution where, from start to finish, we would automate the entire process of designing and manufacturing a molecule. If an LLM could simply give you the answer in seconds, it would be a huge time saving for pharmaceutical companies, “explains Michael Sun, a student graduated from the MIT and co-author of a paper on this technique.
The co-authors of Sun include the main author Gang Liu, a graduate student at the University of Notre Dame; Wojciech Matsik, professor of electrical and computer engineering at MIT who directs the IT design and manufacturing group within the computer intelligence laboratory and artificial intelligence (CSAIL); Meng Jiang, associate professor at the University of Notre Dame; And the main author Jie Chen, principal researcher and director of the Mit-ibm Watson Ai Lab. Research will be presented at the international conference on representations of learning.
Best of both worlds
The models of large languages are not designed to understand the nuances of chemistry, which is one of the reasons why they fight with reverse molecular design, a process of identification of molecular structures which have certain functions or properties.
The LLMS convert the text into representations called tokens, which they use to sequentially predict the following word of a sentence. But molecules are “graphic structures”, made up of atoms and bonds without any particular order, which makes them difficult to code as a sequential text.
On the other hand, AI models based on powerful graphics represent atoms and molecular bonds as nodes and edges interconnected in a graph. Although these models are popular for reverse molecular design, they require complex inputs, cannot understand natural language and give results that can be difficult to interpret.
MIT researchers combined an LLM with AI models based on graphics in a unified setting that draws the best of both worlds.
Llamole, which means a large language model for molecular discovery, uses a basic LLM as a guardian to understand the request of a user – a simple language demand for a molecule with certain properties.
For example, perhaps a user seeks a molecule that can penetrate the blood-brain barrier and inhibit HIV, since it has a molecular weight of 209 and certain liaison characteristics.
As the LLM predicts the text in response to the query, it rocks between the graphic modules.
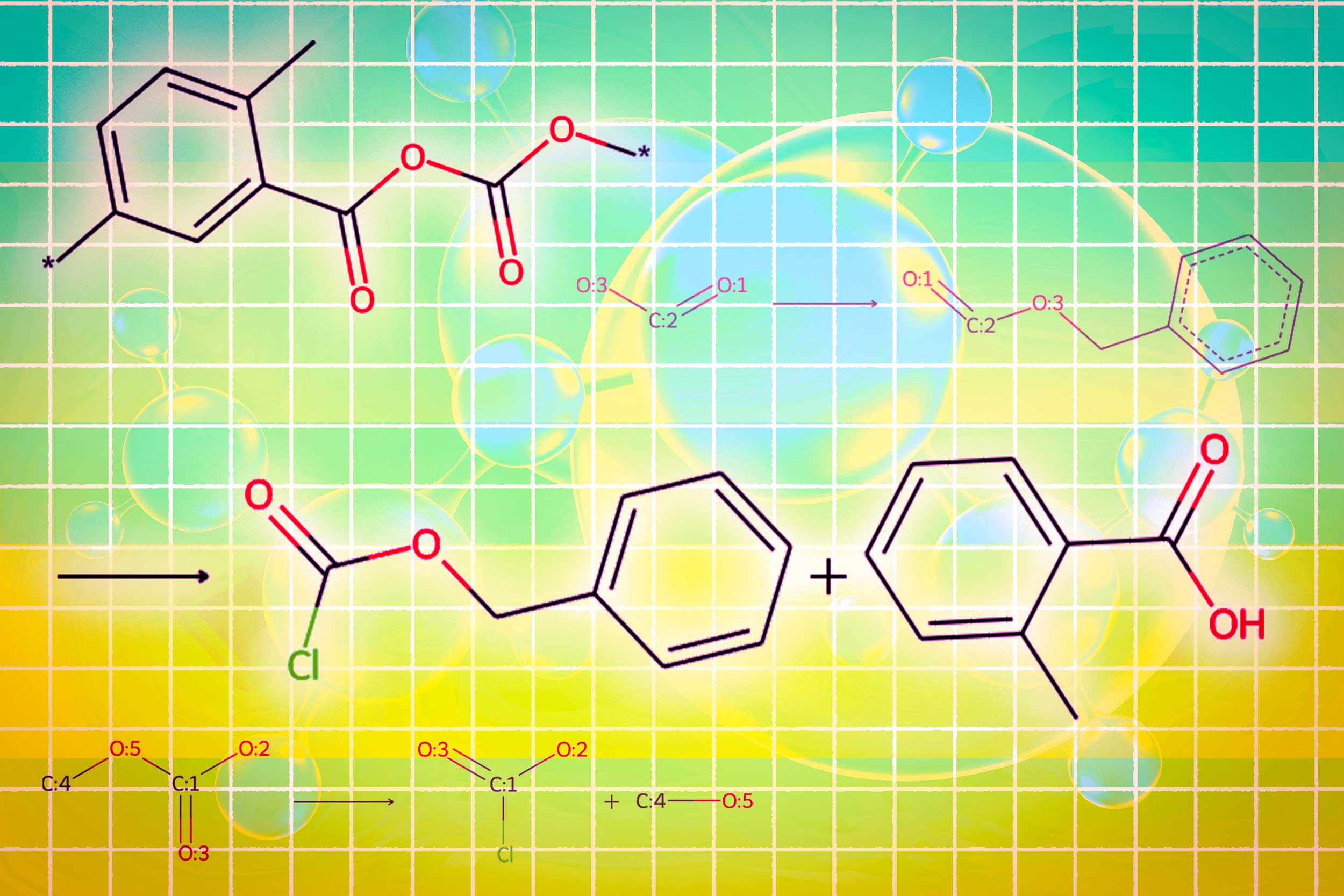
A module uses a graphic diffusion model to generate the conditioned molecular structure with input requirements. A second module uses a network of graphic neurons to code the molecular structure generated in tokens so that LLMS can consume. The final graphic module is a graphic reaction predictor who takes on an intermediate molecular structure as input and predicts a reaction step, looking for the exact set of steps to make the molecule from basic construction blocks.
The researchers have created a new type of trigger token that indicates to the LLM when activating each module. When the LLM predicts a “design” trigger token, it passes to the module which sketches a molecular structure, and when it predicts a “retro” trigger token, it goes to the retrosynthetic planning module which predicts the next reaction step.
“The beauty of this is that all that the LLM generates before activating a particular module is introduced into this module itself. The module learns to function in a consistent way with what preceded, ”says Sun.
In the same way, the release of each module is coded and returned to the LLM generation process, so it understands what each module has done and will continue to predict the tokens according to this data.
Better and simpler molecular structures
In the end, Llamole produces an image of the molecular structure, a textual description of the molecule and a synthetic plan step by step which provides details of how to do so, individual chemical reactions.
In experiments involving the design of molecules that correspond to the user specifications, Llamole has surpassed 10 standard llms, four LLM with fine adjustment and a specific method of the advanced domain. At the same time, he brought the success rate of retrosynthetic planning from 5% to 35% by generating better quality molecules, which means that they had simpler structures and building blocks at a lower cost.
“On their own, LLM find it difficult to understand how to synthesize molecules because it requires a lot of planning in several stages. Our method can generate better molecular structures which are also easier to synthesize, ”explains Liu.
To train and evaluate LMALOLE, the researchers built two data sets from zero, because the existing data sets of the molecular structures did not contain enough details. They have increased hundreds of thousands of patented molecules with natural language descriptions generated by AI and personalized description models.
The data set that they have built to refine the LLM includes models linked to 10 molecular properties, therefore a limitation of the Llamole is that it is formed to design molecules by considering only these 10 digital properties.
In future work, researchers wish to generalize Llamole so that it can incorporate any molecular property. In addition, they plan to improve graph modules to increase the success rate of the Lamole retrosynthesis.
And in the long term, they hope to use this approach to go beyond molecules, creating multimodal LLMs which can manage other types of data based on graphics, such as interconnected sensors in an electric network or transactions on a financial market.
“Llamole demonstrates the feasibility of the use of important language models as an interface to complex data beyond the textual description, and we plan that they are a foundation that interacts with other AI algorithms to solve any graphic problem,” explains Chen.
This research is funded, in part, by the Mit-IBM Watson AI Lab, the National Science Foundation and the Office of Naval Research.